[MR. Park의 건전지] 신약개발 편
생명 연장의 꿈을 실현하는 신약 개발
환자의 생명을 살리기 위한 세 가지 조건이 있습니다. 환자 본인의 의지와 훌륭한 의료진은 물론이며, 무엇보다 적절한 치료제를 사용하는 것이 중요합니다. 신약 개발은 우리가 생명을 연장하기 위해 끊임없이 수행해야 할 과제입니다. 막중한 임무를 띤 신약이 과연 얼마나 엄중한 과정을 거쳐 개발되는지 살펴봅시다.

신약 개발의 중요성은 아무리 강조해도 모자랍니다. 셀 수 없는 전문가들의 거듭된 연구와 확인이 필수적이지요. 신약을 개발하는 과정은 크게 발견과 개발의 두 단계로 나뉘지만, 임상시험 단계까지 나눠보면 매우 다층적입니다. 철저한 신약 개발의 과정을 MR. Park이 친절하게 설명해드리겠습니다!
신약 개발의 첫걸음, 발견(Discovery) 단계
신약 개발의 과정은 발견과 개발의 두 단계로 나뉩니다. 먼저 발견 단계에서는 우선적으로 질병 타깃, 즉 어떠한 질병을 치료하기 위한 목표를 설정하게 됩니다. 타깃이 설정되면 동물시험 등을 거쳐 신약후보물질을 도출합니다. 예를 들어 고혈압 치료제를 개발한다면, 고혈압의 원인을 규명하고 타깃을 선택한 후 기대하는 치료 효과를 발휘할 수 있는 새로운 물질을 만들거나 기존 물질 혹은 수집된 분자 중에서 최적화된 구조를 설계합니다. 이 같은 설계가 끝나면 신약후보물질을 선정하게 됩니다. 가끔 신약 개발 관련 기사에서 접할 수 있는 ‘AB-1234’와 같은 분류체계가 바로 신약후보물질을 일컫는 말입니다.
이 후보물질은 사람에게 임상시험을 하기 전, 전임상 시험을 거칩니다. 실험용 쥐나 족제비, 원숭이 등 여러 종류의 동물들에게 사용해 보고 부작용이나 독성, 유효성 등의 문제점을 살펴본 뒤 임상 시험에 사용할 제제에 대한 제제화 연구를 병행하며 제형 및 처방 등을 결정하는 시험입니다. 최근에는 동물복지 이슈로 전임상 시험에서 동물을 다른 것으로 대체할 방법을 찾고 있습니다.
안전을 위한 임상시험, 개발(Development) 단계
개발 단계에서는 발견 단계에서 확인된 후보물질을 사람에게 사용해 안전성과 유효성을 확인합니다. 우리가 익히 들어 알고 있는 임상시험의 단계입니다. KGCP(의약품임상시험관리기준)에 따르면 임상시험은 개발을 진행하려고 하는 임상시험용의약품의 안전성과 유효성을 증명할 목적으로 해당 약물의 약동(생체에 투여한 약물의 체내 인자에 의한 움직임, 약력(약의 효력), 약리(약에 의하여 일어나는 생체의 생리적 변화) 등의 임상 효과를 확인하고 부작용 등의 이상반응을 조사하기 위해 사람을 통해 시험이나 연구를 실시하는 것입니다. 임상시험의 단계는 다음과 같이 구분됩니다.
[제1상 임상약리상 (Clinical Phamacology)]
먼저 임상시험용 의약품을 최초로 사람에게 투여하는 제1상이 진행됩니다. 소규모(20~100명)의 건강한 지원자를 대상으로 시행하는 단계이며 임상시험용의약품이 사람에게 안전성을 확보할 수 있는지를 확인합니다. 전임상 시험에서 확보된 약리작용 데이터를 근거로 인체에서 약물의 체내 동태(생체에 투여된 약물이 생체 밖으로 배설되기까지의 흡수·분포·대사·배설 거동), 인체에서의 약리작용, 부작용 및 안전한 투여량의 범위 등을 결정하며 가능한 경우 인체 내에서 약리 효과를 탐색합니다.
[제2상 임상연구상(Clinical Investigation)]
제2상은 2a와 2b로 나누어 진행하는 경우가 많습니다. 신약의 안전성과 유효성을 증명하기 위해 잘 통제된 디자인으로 임상을 진행하고 약리 효과를 확인합니다. 적정 용량과 용법을 결정하기 위한 초기 연구를 2a라 하며, 본격적으로 인원을 확대해 진행하는 중추적 연구를 2b라 부릅니다. 일반적으로 면밀하게 평가될 수 있는 인원을 설정해 임상을 실시하며, 보통 100명에서 200명 규모로 진행됩니다. 이 단계에서는 임상시험용 의약품이 환자군에 미치는 치료적 유효성을 탐색해 기대된 작용기전에 따라 진행되는지 검토하고, 의약품을 사용하는 데 최적 용량과 투약 방법을 설정하고자 다양한 정보를 수집하는 치료적 탐색과 분석의 단계입니다. 개발되는 치료제의 성격에 따라 2상 후 승인을 내주는 경우가 있으며 이를 조건부 승인이라고 합니다. 조건부 승인을 받아도 2상까지 확인된 범위 내에서만 처방이 가능하며, 3상을 진행해야 합니다.
[제3상 임상시험상(Clinical Trials)]
제3상은 1,000~5,000명의 대규모 환자를 대상으로 실시합니다. 후보의약품의 안전성과 유효성, 그리고 치료할 수 있는 기대효과 대비 발생할 수 있는 이상반응이나 부작용의 상관관계에 대해 통계적으로 유의미한 데이터를 만들어야 하기 때문입니다. 시험이 진행되는 동안 실생산 규모의 생산계획과 식약청 품목허가를 위한 서류를 준비하는 연구가 동시에 수행됩니다.
[제4상 시판 후 안전성 조사 및 임상시험]
의료·제약업계 종사자가 아니라면 아마 4상을 낯설게 여기는 분들이 많을 겁니다. 흔히 PMS(Post Marketing Surveillance & Post Marketing Clinical Trials)라고 부릅니다. 약의 시판 허가 후 진행되는 4상은 두 갈래로 진행됩니다. 하나는 약물의 이상반응이나 부작용의 빈도에 관해 확실한 추가 정보를 얻는 시판 후 안전성 조사이며, 다른 하나는 3상의 보완적 성격, 이를테면 자료 보완이나 적응증 추가 연구, 특수 환자군 임상 연구 등의 추가 임상연구입니다. PMS를 진행할 시 임상진행 시와 마찬가지로 환자들에게 개인정보동의를 구하고 의료기록과 설문을 통해 개발이 완료된 약물을 한 번 더 점검해 환자들에게 유효하며 안전한 처방이 이루어지고 있는지 확인합니다.
임상시험심사위원회 및 임상시험윤리위원회
임상시험이 과학적, 윤리적으로 정확하게 진행되는지를 지켜보는 기관도 있습니다. 바로 임상시험심사위원회 및 임상시험윤리위원회(IRB, Institutional Review Board)입니다. 연구자는 임상시험의 절차 및 방법에 대한 임상시험계획서와 시험대상자에게 제공할 의약품에 대해 자세한 설명이 들어있는 임상시험자료집, 그리고 시험대상자 동의서와 피해자 보상규약 및 보험, 연구비 내역 등의 서류를 준비해 임상시험심사위원회에 제출하고 심의를 받아야 합니다. 여기에 시험에 참가하는 환자의 권리를 윤리적으로 보호하고 혹시 모를 불상사에 대비하기 위해 5명 이상의 임상시험윤리위원회를 추가로 구성합니다.
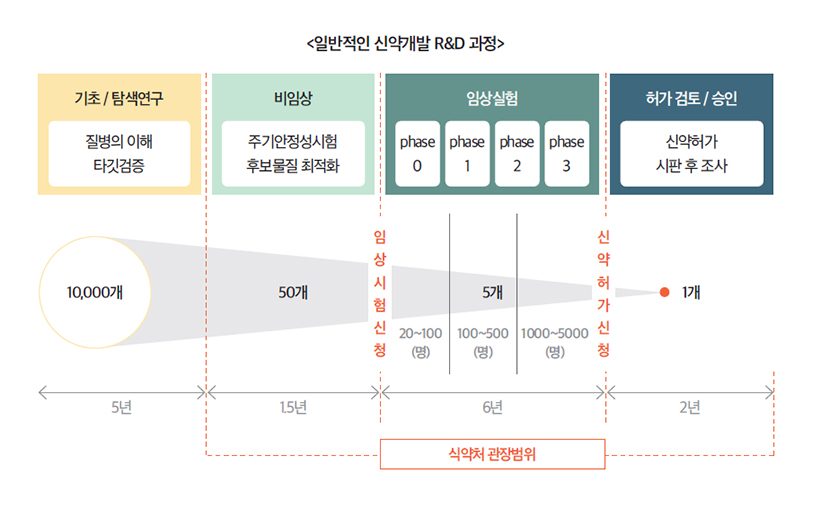
신약 승인은 하늘의 별 따기
유효물질 수준에서 5,000~10,000개의 화합물이 연구개발의 파이프라인으로 들어가지만, 전임상 단계에서 약 50개로 줄고 결국에는 하나만 남아 승인을 받게 됩니다. 국내에서는 지난 1999년 1호 신약 이후 20여 년이 흐른 지금까지 개발신약 허가품목이 단 29개밖에 되지 않으니 얼마나 개발하기 힘든지 짐작할 수 있습니다. 이렇게 어려운 신약 개발 과정을 거쳐 환자가 약을 사용할 수 있을 때까지 10~15년의 연구개발기간이 소요되며, 비용은 100억 원에서 많게는 1조 6천억 원 이상까지도 추정됩니다.
※ 동아약보 2020년 9월호 발췌